THEORETICAL MODELS OF DEFECTS IN REDUCIBLE OXIDES AND AT INTERFACES
WG4 members: Alexander Shluger, Scott Woodley
homepage: www.cmmp.ucl.ac.uk/~kpm/
The activity aims at the developing and application of new theoretical methods for modelling defects and defect properties in reducible oxides and at interfaces with metals and semiconductors for microelectronics and energy applications. The oxides studied include binary oxides MgO, ZnO, SiO2, TiO2, ZrO2, HfO2, as well as more complex oxides, such as CaMnO3 and HfSiO4. We focus on modelling the structure and properties of defects in the bulk and at surfaces of these oxides as well as at interfaces, such as Si/SiO2/HfO2, MgO/Ag, HfO2/TiN and others in the form of ultrathin films and supported nanostructures for microelectronic applications.
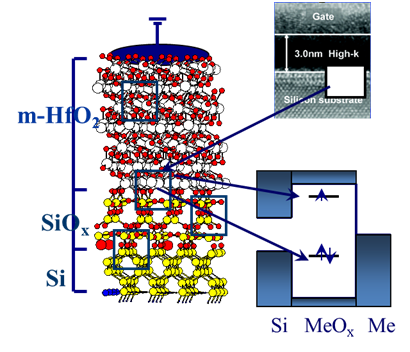 The goal is to achieve the atomic scale understanding of the investigated systems with particular focus on the factors which influence reducibility (i.e. dimensionality, doping, metal support and interaction with supported metal nanoparticles).
The goal is to achieve the atomic scale understanding of the investigated systems with particular focus on the factors which influence reducibility (i.e. dimensionality, doping, metal support and interaction with supported metal nanoparticles).
The research is performed using a combination of atomistic and electronic structure methods. We develop force-fields for atomistic molecular dynamics simulations and effective computational methods for electronic structure calculations, as well as hybrid embedded cluster methods. Computer codes used in simulations include DL-Poly, LAMMPS, Guess, CRYSTAL09, CP2K, VASP and others.
