
Quantum chemical simulations on reactions at surfaces of reducible oxides
Institute of Chemistry, Humboldt University Berlin, Germany
Dr. Joachim Paier received his PhD in Physics at the University of Vienna, Austria in 2008. After two postdoctoral stays with Prof. Dr. Georg Kresse, University of Vienna, Austria in 2008 and with Prof. Gustavo E. Scuseria, Rice University, Houston, TX, USA in 2009, he joined the group of Prof. Dr. Joachim Sauer at the Humboldt University Berlin as a research associate (2010). Besides of his teaching responsibilities, he is designing and supervising the research projects targeting at atomistic simulations on selective oxidation reactions at transition metal oxide catalysts supported on rare-earth metal oxide surfaces.
Reducible oxides are the major components of many present-day catalysts widely applied in chemical industry for selective oxidation reactions. A valid example is the oxidative dehydrogenation (ODH) of lower alkanes and alcohols, as e.g. propane oxidation to propene or partial oxidation of methanol to formaldehyde. However, in order to optimize turnover frequencies, minimize byproduct formation, or ensure reasonable lifetimes, atomic level insights into possible reaction mechanisms at the catalyst’s surface are mandatory. Detailed mechanistic understanding in terms of geometric structure (How does the active site look like under various oxygen partial pressures?) as well as electronic structure (Are charge-transfer processes important?) can be generated by quantum chemical simulations.

Figure 1: Dissociative adsorption of CH3OH from the gas phase at the (pseudo-) O-vacancy formed after relaxation of a surface O atom; subsequent relaxation of the surface, and H-transfer to the next-neighbor, V-anchoring oxygen atom. Simulations use periodic PBE+U together with nudged-elastic band algorithms. Courtesy of Thomas Kropp.
At the Humboldt University Berlin, we address these questions by periodic density functional theory (DFT) calculations (among other quantum chemical techniques). Figure 1 shows a series of structures along a minimum energy pathway of the ODH of methanol at VO2 supported on a CeO2(111) surface. To locate transition state structures we use a two-step procedure involving (i) generation of a reasonable guess for the TS within a nudged-elastic band run, and (ii) refinement calculations using more sophisticated techniques, such as climbing image or improved dimer methods.
DFT is the workhorse when it comes to simulations on extended systems. However, its incidentally non-systematic methodic accuracy may hamper the accurate description of defective oxides (point defects or aliovalent dopants). Hence, are relative energies or activation barriers (corrected for zero-temperature effects and thermal contributions) reliable enough?
It is well known, that shortcomings of DFT within the formulation of Kohn and Sham due to the approximate nature of the functionals used may spoil results. For instance, lack of a correct description of dispersion interactions or inclusion of electron self-interaction (i.e., self-interaction error; SIE) favoring over-delocalization of electronic states may render accurate predictions elusive. Therefore, we make strong efforts to acquire knowledge on reliability of less compute intensive methods (e.g. DFT+U) and compare results with those of more sophisticated, but also more compute intensive approaches. One option beyond conventional LDA/GGA approximations is the application of hybrid functionals. Hybrid functionals admix a certain fraction of the non-local Fock exchange to the LDA/GGA exchange energy and therefore partially rectify the SIE. However, the amount of admixed Fock exchange appears to depend on the screening properties of the system, i.e. it needs to be considered as a semi-empirical parameter.
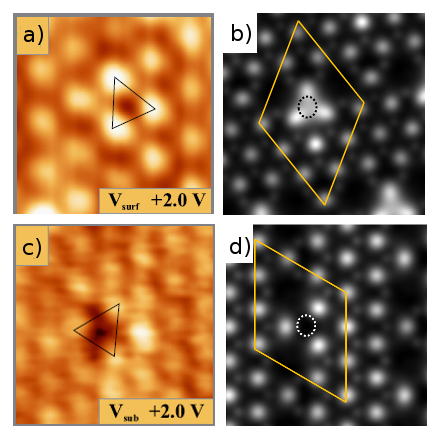
Figure 2: a) and b) represent the respective experimental and simulated (Tersoff-Hamann approximation) STM empty state image of the oxygen surface vacancy at a positive bias voltage of 2 eV. Similar images are given for the subsurface vacancy in c) and d).
As an example for the application of hybrids to real systems, Figures 2 and 3 highlight some of our most recent findings on the adsorption of Au atoms on a partially reduced CeO2(111) surface [Y. Pan, N. Nilius, H.-J. Freund, J. Paier, C. Penschke, and J. Sauer, Phys. Rev. Lett. (2013); in print]. We found that neither LDA+U nor PBE+U predict the correct adsorption site for a Au atom near a subsurface oxygen vacancy. However, hybrid functionals predict adsorption of Au atop a Ce3+ ion (Fig. 3b) as well as creation of Au– ions, i.e. charge transfer from Ce3+ to Au. Given the rather large ionic radius of Ce3+ compared to Ce4+ (1.01 versus 0.87 Å), lattice strain builds up upon reduction of the oxide film. This strain is released, which in turn stabilizes the Au-Ce interaction, when the electron from Ce3+ occupying a 4f orbital is transferred into the Au 6s state. This scenario is in perfect agreement with images obtained using STM experiments accomplished at the FHI Berlin by the group of Nilius/Freund. These images show the formation of distinct Au pairs at a reduced ceria film.
Experimentally important, discriminating surface O-vacancies (three bright maxima) and subsurface oxygen vacancies by empty state STM is possible and clearly supported by theory (Fig. 2 b and d, using the Tersoff-Hamann approximation). Differences are easily explained when considering the electron-tunnelling paths that are responsible for the observed contrast. At positive bias, electrons are injected into empty states of the surface that are mainly localized at the Ce ions and render the Ce sub-lattice visible in the images. On the ideal surface, these Ce 4f states are highly compressed due to the oxygen layer above and therefore less accessible for the tunnelling electrons. In presence of a surface O defect, the 4f orbitals are able to spill out into the void, which enables at better overlap with the tip wave functions [cf. Jerratsch et al., Phys. Rev. Lett. 106, 246801 (2011)]. As a result, the Ce4+ ions next to a surface vacancy turn bright while the Ce3+ ions remain darker as their 4f manifold is shifted to higher energy and generally less accessible. The effect vanishes in empty state images of a subsurface oxygen vacancy, where no hole exists in the surface and tunnelling into the Ce 4f states remains hampered by the surface O layer. The only contrast change in this case is a weak depression that marks the defect position and originates from a slight reconfiguration of the Ce electronic states in response to the subsurface vacancy.
![Figure 3: (a) Distance histogram determined for 150 Au-pairs on the ceria surface. (b) Relative energies [eV] obtained using HSE (optimized) and B3LYP (single point) for three relevant Au adsorption sites (Values in parenthesis include dispersion effects). (c) Scheme of a p(44) unit cell of CeO2(111). The subsurface O vacancy, Vsub, is indicated by a dashed circle. The excess electrons occupy preferentially two Ce-sites in the 2nd coordination shell of Vsub (here: b1, b2, or b3). Published in Pan, Nilius, Freund, Paier, Penschke, Sauer, Phys. Rev. Lett (2013).](http://www.cost-redox.nano.cnr.it/wp-content/uploads/2013/10/Figure-3.png)
Figure 3: (a) Distance histogram determined for ~150 Au-pairs on the ceria surface. (b) Relative energies [eV] obtained using HSE (optimized) and B3LYP (single point) for three relevant Au adsorption sites (Values in parenthesis include dispersion effects). (c) Scheme of a p(4×4) unit cell of CeO2(111). The subsurface O vacancy, Vsub, is indicated by a dashed circle. The excess electrons occupy preferentially two Ce-sites in the 2nd coordination shell of Vsub (here: b1, b2, or b3). Published in Pan, Nilius, Freund, Paier, Penschke, Sauer, Phys. Rev. Lett. (2013).
The basic question raised by the STM observations targeted at reasons for formation of Au pairs separated by a distance of two lattice constants of the surface unit cell. Figure 3(a) presents the above mentioned experimental findings showing the relative abundance of Au pairs at various interatomic distances. There is a clear maximum at 7.6 Å, which corresponds to twice the length of the CeO2(111) lattice parameter. The STM puzzle could be solved by simulations using the screened hybrid functional HSE by Heyd, Scuseria, and Ernzerhof, however we emphasize that the results do not critically depend on the exact type of functional, as demonstrated by single point calculations using B3LYP (Fig. 3b).
To further support the Ce3+ titration scenario, we compared the experimentally determined Au-Au distances with the equilibrium separation of the two Ce3+ created by a single O subsurface defect. Upon creation of a subsurface vacancy, the two electrons left behind localize in Ce ions sitting in the second coordination shell with respect to the vacancy (b1, b2, b3 sites, Fig. 3b). Although simulations predict a rather small energy difference of about 250 to 400 meV comparing the calculated energy minimum with other Ce3+ pair configurations, we consider this energy gap as a definite thermodynamic driving force. Formation of two Ce3+ ions in nearest-neighbor position to the vacancy is found to be almost 500 meV uphill in energy relative to the minimum energy configuration. This implies a most favorable Ce3+ pair distance of 7.6 Å.
Titration of reduced Ce sites by Au adatoms is a new and exciting result. As a consequence, Au can be used as a marker for Ce3+ ions, providing unique information on electron-localization phenomena in reduced ceria. In spite of this unprecedented outcome, further work is however needed to e.g. pinpoint a critical defect concentration (i.e. level of surface strain) impelling charge-transfer from Ce to Au, which as kind of a valve for strain relief drives the titration of Ce3+ by Au atoms.
